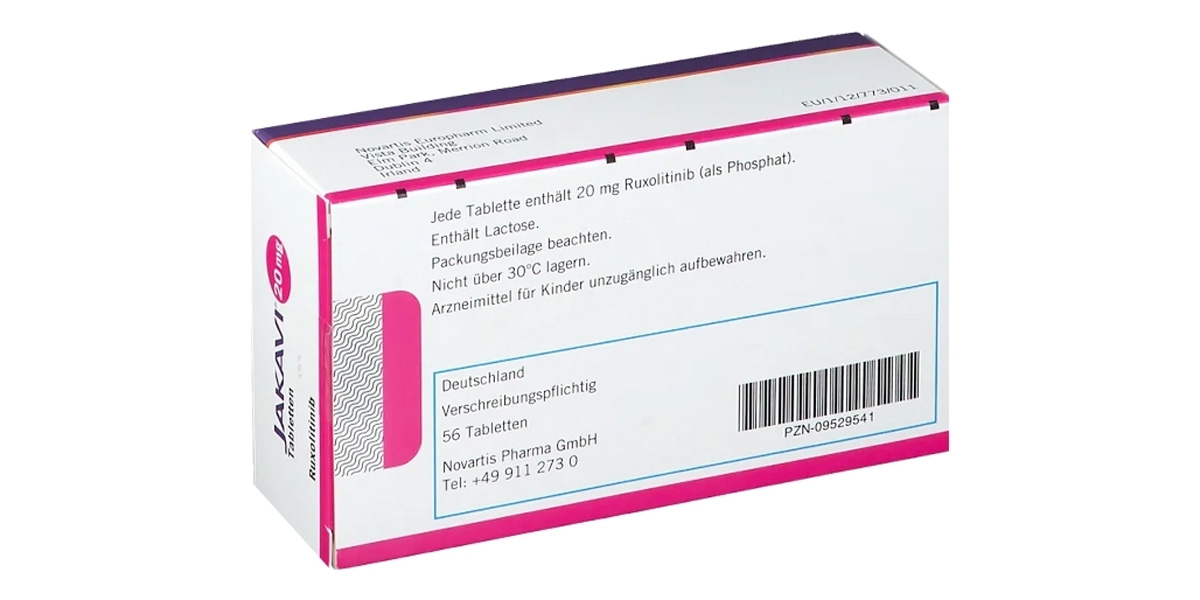
1. Thành phần
Thông tin thành phần | Hàm lượng |
Ruxolitinib | 20 mg |
2. Chỉ định
Jakavi được chỉ định để điều trị bệnh nhân bị xơ tủy xương , bao gồm xơ tủy nguyên phát, xơ tủy sau khi bị tăng hồng cầu vô căn hoặc xơ tủy sau khi bị tăng tiểu cầu vô căn.
3. Cách dùng
– Cách dùng
Jakavi được dùng đường uống và có thể dùng cùng hoặc không cùng với thức ăn.
Điều trị với Jakavi chỉ nên được tiến hành bởi bác sĩ có kinh nghiệm sử dụng thuốc điều trị ung thư.
Hướng dẫn theo dõi
Công thức máu toàn phần: Phải xét nghiệm công thức máu toàn phần trước khi khởi đầu điều trị bằng Jakavi.
Cần theo dõi công thức máu toàn phần mỗi 2 – 4 tuần cho đến khi liều dùng ổn định và sau đó được chỉ định theo lâm sàng.
– Liều dùng
Liều khởi đầu
Liều khởi đầu khuyến cáo của Jakavi là 15 mg, dùng đường uống 2 lần/ngày đối với những bệnh nhân có số lượng tiểu cầu từ 100.000 – 200.000/mm3 và 20 mg, 2 lần/ngày đối với những bệnh nhân có số lượng tiểu cầu > 200.000/mm3. Thông tin còn hạn chế về việc khuyến cáo liều khởi đầu cho những bệnh nhân có số lượng tiểu cầu từ 50.000/mm3 – 100.000/mm3. Liều khởi đầu tối đa được khuyến cáo ở những bệnh nhân này là 5 mg, 2 lần/ngày và các bệnh nhân này nên được chỉnh liều một cách thận trọng.
Điều chỉnh liều
Liều dùng nên được điều chỉnh dựa trên độ an toàn và hiệu quả. Nên tạm ngưng điều trị khi số lượng tiểu cầu dưới 50.000/mm3 hoặc số lượng bạch cầu trung tính tuyệt đối dưới 500/mm3. Sau khi số lượng huyết cầu phục hồi lên cao hơn những mức này, có thể bắt đầu dùng thuốc trở lại với liều 5 mg, 2 lần/ngày và tăng dần dựa trên việc theo dõi cẩn thận công thức máu toàn phần.
Nên xem xét giảm liều nếu số lượng tiểu cầu giảm dưới 100.000/mm3 với mục đích tránh phải gián đoạn liều dùng vì giảm tiểu cầu.
Nếu hiệu quả được xem là chưa đạt và số lượng huyết cầu đầy đủ thì có thể tăng liều lên tối đa thêm 5 mg, 2 lần/ngày; lên đến liều tối đa 25 mg, 2 lần/ngày.
Không nên tăng liều khởi đầu trong vòng 4 tuần điều trị đầu tiên và sau đó nếu cần tăng thì phải chờ ít nhất mỗi 2 tuần mới tăng một lần.
Hướng dẫn sử dụng
Liều tối đa của Jakavi là 25 mg, 2 lần/ngày.
Nếu quên một liều, bệnh nhân không nên uống một liều bổ sung mà nên dùng liều kế tiếp theo thường lệ được kê đơn.
Có thể tiếp tục điều trị chừng nào lợi ích vẫn còn nhiều hơn nguy cơ.
Điều chỉnh liều khi dùng đồng thời với thuốc ức chế mạnh CYP3A4 hoặc fluconazol
Khi Jakavi được dùng với thuốc ức chế mạnh CYP3A4 hoặc thuốc ức chế kép CYP2C9 và CYP3A4 (ví dụ: Fluconazol), nên giảm tổng liều hàng ngày của Jakavi xuống khoảng 50% bằng cách giảm liều dùng 2 lần/ngày hoặc giảm số lần dùng thuốc xuống còn 1 lần/ngày với liều tương ứng khi liều dùng 2 lần/ngày không khả thi. Tránh dùng đồng thời Jakavi va fluconazol với liều lớn hơn 200 mg/ngày.
Khuyến cáo theo dõi thường xuyên hơn các thông số huyết học và các dấu hiệu, triệu chứng lâm sàng của các phản ứng phụ liên quan với Jakavi khi bắt đầu dùng một thuốc ức chế mạnh CYP3A4 hoặc thuốc ức chế kép CYP2C9 và CYP3A4.
Các nhóm bệnh nhân đặc biệt
- Suy thận:
Không cần điều chỉnh liều đặc biệt ở bệnh nhân suy thận nhẹ hoặc trung bình.
Ở bệnh nhân bị suy thận nặng (độ thanh thải creatinin dưới 30 ml/phút), liều khởi đầu khuyến cáo dựa trên số lượng tiểu cầu đối với bệnh nhân bị xơ tủy xương (MF) nên được giảm xuống khoảng 50%, dùng 2 lần/ngày. Cần theo dõi bệnh nhân cẩn thận về độ an toàn và hiệu quả trong khi điều trị bằng Jakavi.
Dữ liệu còn hạn chế trong việc xác định các lựa chọn liều dùng tốt nhất cho bệnh nhân bị bệnh thận giai đoạn cuối (ESRD) đang được lọc thận. Các mô phỏng về dược động học/dược lực học dựa trên dữ liệu hiện có ở nhóm bệnh nhân này cho thấy liều khởi đầu đối với bệnh nhân bị xơ tủy xương (MF) có bệnh thận giai đoạn cuối đang được lọc thận là một liều đơn 15 – 20 mg hoặc 2 liều 10 mg dùng cách nhau 12 giờ, được dùng sau khi lọc thận và chỉ dùng vào ngày lọc thận. Khuyến cáo dùng một liều đơn 15 mg đối với bệnh nhân bị xơ tủy xương có số lượng tiểu cầu từ 100.000/mm3 – 200.000/mm3. Khuyến cáo dùng một liều đơn 20 mg hoặc 2 liều 10 mg cách nhau 12 giờ đối với bệnh nhân bị xơ tủy xương có số lượng tiểu cầu > 200.000/mm3. Các liều kế tiếp (liều đơn hoặc 2 liều 10 mg dùng cách nhau 12 giờ) chỉ nên dùng vào những ngày lọc thận sau mỗi lần lọc thận.
- Suy gan:
Ở bệnh nhân có bất kỳ mức độ suy gan nào, liều khởi đầu khuyến cáo dựa trên số lượng tiểu cầu nên được giảm xuống khoảng 50%, dùng 2 lần/ngày. Cần điều chỉnh các liều kế tiếp dựa trên việc theo dõi cẩn thận về độ an toàn và hiệu quả. Những bệnh nhân được chẩn đoán suy gan trong khi điều trị bằng Jakavi cần được xét nghiệm công thức máu toàn bộ, bao gồm cả tỷ lệ bạch cầu, theo dõi ít nhất mỗi 1 – 2 tuần trong 6 tuần đầu tiên sau khi bắt đầu điều trị bằng Jakavi và sau đó khi được chỉ định trên lâm sàng một khi chức năng gan và số lượng huyết cầu đã ổn định. Liều Jakavi có thể được điều chỉnh để làm hạ thấp nguy cơ giảm huyết cầu.
- Bệnh nhi:
Độ an toàn và hiệu quả của Jakavi ở bệnh nhi chưa được xác định.
Bệnh nhân cao tuổi:
Không có sự điều chỉnh liều bổ sung nào được khuyến cáo đối với bệnh nhân cao tuổi.
- Ngừng điều trị:
Có thể tiếp tục điều trị chừng nào lợi ích vẫn lớn hơn nguy cơ. Tuy nhiên, nên ngừng thuốc sau 6 tháng nếu không có sự giảm kích thước lách hoặc không có cải thiện triệu chứng kể từ khi bắt đầu điều trị.
Đối với những bệnh nhân cho thấy có một số cải thiện nhất định trên lâm sàng, khuyến cáo nên ngừng điều trị bằng ruxolitinib nếu họ vẫn bị tăng chiều dài lách 40% so với kích thước ban đầu (gần tương đương với một mức tăng 25% về thể tích lách) và không có sự cải thiện rõ rệt các triệu chứng liên quan đến bệnh.
– Quá liều
Chưa có thuốc giải độc đối với trường hợp quá liều Jakavi. Các liều đơn lên đến 200 mg đã được dùng với khả năng dung nạp cấp chấp nhận được. Các liều lặp lại cao hơn so với liều được khuyến cáo có liên quan với tăng ức chế tủy xương bao gồm giảm bạch cầu, thiếu máu và giảm tiểu cầu. Cần tiến hành điều trị hỗ trợ thích hợp.
Thẩm phân máu khó có thể làm tăng thải trừ Jakavi.
4. Chống chỉ định
- Quá mẫn với hoạt chất hoặc với bẤt kỳ tá dược nào của thuốc.
- Phụ nữ có thai và cho con bú.
5. Tác dụng phụ
Thông báo cho bác sỹ những tác dụng không mong muốn gặp phải khi sử dụng thuốc.
Tóm tắt về đặc tính an toàn
Đánh giá về độ an toàn được dựa trên tổng số 855 bệnh nhân (bị xơ tủy xương hoặc một chỉ định khác trên thực nghiệm) được điều trị bằng Jakavi trong các nghiên cứu pha 2 và pha 3.
Trong thời gian chọn điều trị ngẫu nhiên của hai nghiên cứu then chốt COMFORT-I và COMFORT-II, các bệnh nhân đã có thời gian sử dụng Jakavi trung bình là 10,8 tháng (trong khoảng 0,3 – 23,5 tháng). Đa số bệnh nhân (68,4%) đã được điều trị trong ít nhất 9 tháng. Trong số 301 bệnh nhân, 111 bệnh nhân (36,9%) có số lượng tiểu cầu lúc ban đầu từ 100.000/mm3 – 200.000/mm3 và 190 bệnh nhân (63,1%) có số lượng tiểu cầu lúc ban đầu > 200.000/mm3.
Trong các nghiên cứu lâm sàng này, ngừng thuốc do các phản ứng bất lợi bất kể quan hệ nhân quả đã được quan sát thấy ở 11,3% bệnh nhân.
Các phản ứng bất lợi của thuốc được báo cáo thường gặp nhất là giảm tiểu cầu và thiếu máu.
Các phản ứng bất lợi về huyết học (bất kỳ cấp độ nào theo Tiêu chuẩn thuật ngữ chung về các phản ứng bất lợi (CTCAE)) bao gồm thiếu máu (82,4%), giảm tiểu cầu (69,8%) và giảm bạch cầu trung tính (16,6%).
Thiếu máu, giảm tiểu cầu và giảm bạch cầu trung tính là các tác dụng bất lợi liên quan với liều dùng.
Ba phản ứng bất lợi không phải huyết học thường gặp nhất là bầm tím (21,6%), xây xẩm (15,3%) và nhức đầu (14,0%).
Ba bất thường về xét nghiệm không phải huyết học thường gặp nhất là tăng alanin aminotransferase (27,2%), tăng aspartat aminotransferase (19,9%) và tăng cholesterol máu (16,9%).
Độ an toàn lâu dài: Như được dự kiến với thời gian theo dõi kéo dài, tần suất tích lũy của một số phản ứng bất lợi tăng lên trong đánh giá của dữ liệu an toàn theo dõi 3 năm (thời gian sử dụng thuốc trung vị là 33,4 tháng trong nghiên cứu COMFORT-I và COMFORT- II đối với những bệnh nhân được chọn ngẫu nhiên ban đầu để điều trị bằng ruxolitinib) từ 457 bệnh nhân bị xơ tủy xương được điều trị bằng ruxolitinib trong thời gian chọn ngẫu nhiên và giai đoạn điều trị mở rộng của 2 nghiên cứu pha 3 then chốt. Việc đánh giá này bao gồm dữ liệu từ những bệnh nhân được chọn ngẫu nhiên ban đầu để điều trị bằng ruxolitinib (n = 301) và những bệnh nhân đã được điều trị bằng ruxolitinib sau khi chuyển nhóm từ các nhóm điều trị đối chứng (n = 156). Với những dữ liệu cập nhật này, ngừng điều trị do các phản ứng bất lợi đã được quan sát thấy ở 21,4% bệnh nhân điều trị bằng ruxolitinib.
Bảng tóm tắt các phản ứng bất lợi của thuốc từ các thử nghiệm lâm sàng
Phản ứng bất lợi của thuốc từ các thử nghiệm lâm sàng (Bảng 1) được liệt kê theo nhóm hệ cơ quan của MedDRA. Trong mỗi nhóm hệ cơ quan, các phản ứng bất lợi của thuốc được sắp xếp theo tần suất, đầu tiên là các phản ứng thường gặp nhất. Ngoài ra, loại tần suất tương ứng đối với mỗi phản ứng bất lợi được dựa trên quy ước sau đây (CIOMS III): rất thường gặp (≥ 1/10); thường gặp (≥ 1/100 đến < 1/10); ít gặp (≥ 1/1.000 đến < 1/100); hiếm gặp (≥ 1/10.000 đến < 1/1.000); rất hiếm gặp (< 1/10.000).
Trong các chương trình nghiên cứu lâm sàng, mức độ nặng của các phản ứng bất lợi của thuốc được đánh giá dựa trên Tiêu chuẩn thuật ngữ chung về các phản ứng bất lợi (CTCAE) xác định độ 1 = nhẹ, độ 2 = trung bình, độ 3 = nặng và độ 4 = đe dọa tính mạng hoặc gây tàn tật.
Bảng 1. Báo cáo nhóm tần suất của các phản ứng bất lợi của thuốc được báo cáo trong các nghiên cứu pha 3 (COMFORT-I, COMFORT-II)
Phản ứng bất lợi của thuốc và độ CTCAE | Nhóm tần suất đối với bệnh nhân bị xơ hóa tủy xương (MF) |
Nhiễm trùng và nhiễm ký sinh trùng | |
Nhiễm trùng đường tiết niệu | Rất thường gặp |
Bệnh zona (Herpes zoster) | Thường gặp |
Bệnh lao | Ít gặp |
Rối loạn máu và hệ bạch huyết | |
Thiếu máu | – |
Độ 4 theo CTCAE (< 6,5 g/dl) | Rất thường gặp |
Độ 3 theo CTCAE (< 8,0 – 6,5 g/dl) | Rất thường gặp |
Bất kỳ cấp độ nào theo CTCAE | Rất thường gặp |
Giảm tiểu cầu | – |
Độ 4 theo CTCAE (< 25.000/mm3) | Thường gặp |
Độ 3 theo CTCAE (< 8,0 – 6,5 g/dl) | Thường gặp |
Bất kỳ cấp độ nào theo CTCAE | Rất thường gặp |
Giảm bạch cầu trung tính | – |
Độ 4 theo CTCAE (< 500/mm3) | Thường gặp |
Độ 3 theo CTCAE (< 1.000 – 500/mm3) | Thường gặp |
Bất kỳ cấp độ nào theo CTCAE | Rất thường gặp |
Rối loạn chuyển hóa và dinh dưỡng | |
Tăng cân | Rất thường gặp |
Tăng cholesterol máu | Rất thường gặp |
Rối loạn hệ thần kinh | |
Xây Xẩm | Rất thường gặp |
Nhức đầu | Rất thường gặp |
Rối loạn tiêu hóa | |
Đầy hơi | Thường gặp |
Rối loạn gan mật | |
Tăng alanin aminotransferase | – |
Độ 3 theo CTCAE | Thường gặp |
Bất kỳ cấp độ nào theo CTCAE | Rất thường gặp |
Tăng aspartat aminotransferase | – |
Bất kỳ cấp độ nào theo CTCAE |
|
Rối loạn da và mô dưới da | |
Bầm tím | Rất thường gặp |
Sau khi ngừng thuốc, bệnh nhân có thể bị các triệu chứng xơ tủy trở lại như mệt mỏi, đau xương, sốt, ngứa, ra mồ hôi ban đêm, lách to có triệu chứng và giảm cân. Trong các nghiên cứu lâm sàng, tổng điểm số triệu chứng về các triệu chứng xơ tủy dần dần trở về trị số ban đầu trong vòng 7 ngày sau khi ngừng thuốc.
Trong một chỉ định khác đang nghiên cứu có quan sát thấy thêm các phản ứng bất lợi sau đây: Tăng huyết áp (thường gặp), táo bón (thường gặp), tăng triglycerid máu độ 1 theo CTCAE (rất thường gặp).
Mô tả các phản ứng phụ của thuốc chọn lọc
- Thiếu máu
Trong các nghiên cứu lâm sàng pha 3, thời gian trung vị dẫn đến khởi phát thiếu máu lần đầu tiên độ 2 trở lên theo CTCAE là 1,5 tháng. Một bệnh nhân (0,3%) đã ngừng điều trị do thiếu máu.
Ở những bệnh nhân đang điều trị bằng Jakavi, mức giảm trung bình về hemoglobin đạt đến mức thấp nhất khoảng 15 – 20 g/l dưới mức ban đầu sau 8 – 12 tuần điều trị và sau đó phục hồi dần dần để đạt được một trạng thái ổn định mới khoảng 10 g/l dưới mức ban đầu. Hiện tượng này được quan sát thấy ở bệnh nhân bất kể họ có được truyền máu trong khi điều trị hay không.
Trong một nghiên cứu ngẫu nhiên, có đối chứng với giả dược (COMFORT-I), 59,4% bệnh nhân được điều trị bằng Jakavi và 37,1% bệnh nhân được điều trị bằng giả dược được truyền hồng cầu trong khi điều trị. Trong nghiên cứu COMFORT-II, tỷ lệ truyền hồng cầu lắng là 51,4% ở nhóm dùng Jakavi và 38,4% ở nhóm dùng trị liệu tốt nhất hiện có.
- Giảm tiểu cầu
Trong các nghiên cứu lâm sàng pha 3, ở những bệnh nhân phát sinh giảm tiểu cầu độ 3 hoặc 4, thời gian trung vị dẫn đến khởi phát khoảng 8 tuần. Giảm tiểu cầu thường có hồi phục khi giảm liều hoặc tạm ngừng dùng thuốc. Thời gian trung vị đến khi phục hồi số lượng tiểu cầu trên 50.000/mm3 là 14 ngày. Truyền tiểu cầu đã được sử dụng trong suốt giai đoạn ngẫu nhiện đối với 4,5% bệnh nhân đang dùng Jakavi và 5,8% bệnh nhân được điều trị đối chứng. Ngừng điều trị do giảm tiểu cầu xảy ra ở 0,7% bệnh nhân đang dùng Jakavi và 0,9% bệnh nhân điều trị đối chứng. Những bệnh nhân có số lượng tiểu cầu 100.000/mm3 – 200.000/mm3 trước khi bắt đầu dùng Jakavi có tần suất giảm tiểu cầu độ 3 hoặc 4 cao hơn so với những bệnh nhân có số lượng tiểu cầu > 200.000/mm3 (64,2% so với 35,4%).
- Giảm bạch cầu trung tính
Trong các nghiên cứu lâm sàng pha 3, ở những bệnh nhân phát sinh giảm bạch cầu trung tính độ 3 hoặc 4, thời gian trung vị dẫn đến khởi phát là 12 tuần. Tạm ngưng hoặc giảm liều do giảm bạch cầu trung tính trong suốt giai đoạn ngẫu nhiên của nghiên cứu đã được báo cáo ở 1,3% bệnh nhân và 0,3% bệnh nhân đã ngừng hẳn điều trị do giảm bạch cầu trung tính.
- Nhiễm trùng đường tiết niệu
Trong các nghiên cứu lâm sàng pha 3, nhiễm trùng đường tiết niệu độ 3 hoặc 4 đã được báo cáo ở 1,0% bệnh nhân. Nhiễm khuẩn huyết có nguồn gốc tiết niệu được báo cáo ở 1,0% bệnh nhân và nhiễm trùng thận ở 1 bệnh nhân.
- Bệnh zona (Herpes zoster)
Trong các nghiên cứu lâm sàng pha 3, bệnh zona (herpes zoster) độ 3 hoặc 4 đã được báo cáo ở 1 bệnh nhân.
6. Lưu ý |
– Thận trọng khi sử dụngĐọc kỹ hướng dẫn sử dụng trước khi dùng. Nếu cần thêm thông tin, xin hỏi ý kiến bác sĩ. Thuốc này chỉ dùng theo sự kê đơn của bác sĩ. Giảm số lượng tế bào máu: Điều trị bằng Jakavi có thể gây ra các phản ứng phụ về huyết học, bao gồm giảm tiểu cầu, thiếu máu và giảm bạch cầu trung tính. Phải xét nghiệm công thức máu toàn phần trước khi bắt đầu điều trị bằng Jakavi. Ngừng điều trị ở các bệnh nhân có số lượng tiểu cầu dưới 50.000/mm3 hoặc số lượng bạch cầu trung tính tuyệt đối dưới 500/mm3. Người ta đã quan sát thấy rằng những bệnh nhân có số lượng tiểu cầu thấp (< 200.000/mm3) vào lúc bắt đầu điều trị dễ bị giảm tiểu cầu hơn trong thời gian điều trị. Giảm tiểu cầu thường hồi phục được và thường được xử trí bằng cách giảm liều hoặc tạm ngừng sử dụng Jakavi. Tuy nhiên, có thể cần phải truyền tiểu cầu khi được chỉ định trên lâm sàng. Những bệnh nhân phát sinh thiếu máu có thể cần phải truyền máu. Cũng có thể cần phải xem xét điều chỉnh liều đối với bệnh nhân bị thiếu máu. Những bệnh nhân có lượng hemoglobin dưới 10,0 g/dl lúc bắt đầu điều trị có nguy cơ cao hơn bị giảm hemoglobin dưới 8,0 g/dl trong khi điều trị so với những bệnh nhân có hemoglobin lúc ban đầu cao hơn (79,3% so với 30,1%). Khuyến cáo nên theo dõi thường xuyên hơn về các thông số huyết học, các dấu hiệu và triệu chứng lâm sàng của những phản ứng bất lợi liên quan đến Jakavi đối với những bệnh nhân có hemoglobin ban đầu dưới 10,0 g/dl. Giảm bạch cầu trung tính (Số lượng bạch cầu trung tính tuyệt đối (ANC) < 500/mm3) thường hồi phục được và thường được xử trí bằng cách tạm ngừng sử dụng Jakavi. Cần theo dõi công thức máu toàn phần khi được chỉ định trên lâm sàng và điều chỉnh liều khi cần thiết. Nhiễm trùng: Cần đánh giá bệnh nhân về nguy cơ phát sinh nhiễm khuẩn, nhiễm mycobacterium và nhiễm virus nghiêm trọng. Mắc bệnh lao đã được báo cáo ở bệnh nhân đang dùng Jakavi để điều trị xơ tủy. Cần lưu ý về khả năng bị bệnh lao tiềm ẩn hoặc dạng hoạt động. Không nên bắt đầu điều trị bằng Jakavi cho đến khi các nhiễm trùng nghiêm trọng dạng hoạt động đã được giải quyết. Bác sĩ nên theo dõi cẩn thận những bệnh nhân đang được điều trị bằng Jakavi về các dấu hiệu và triệu chứng của nhiễm trùng và bắt đầu điều trị thích hợp ngay lập tức. Đã có báo cáo về tải lượng virus viêm gan B (nồng độ HBV-DNA) tăng lên, có và không có tăng kèm theo alanin aminotransferase và aspartat aminotransferase ở những bệnh nhân bị nhiễm HBV mạn tính đang dùng Jakavi. Chưa rõ ảnh hưởng của Jakavi đối với sự tăng sinh virus ở bệnh nhân bị nhiễm HBV mạn tính. Bệnh nhân bị nhiễm HBV mạn tính cần được điều trị và theo dõi theo các hướng dẫn lâm sàng. Bệnh zona (Herpes Zoster): Bác sĩ nên giáo dục cho bệnh nhân về các dấu hiệu và triệu chứng sớm của bệnh zona (herpes zoster) và khuyên họ nên đi điều trị càng sớm càng tốt. Bệnh chất trắng não đa ổ tiến triển: Bệnh chất trắng não đa ổ tiến triển (PML) đã được báo cáo với việc điều trị xơ tủy bằng ruxolitinib. Bác sĩ nên cảnh giác với các triệu chứng thần kinh – tâm thần gợi ý bệnh chất trắng não đa ổ tiến triển. Ung thư da không phải u hắc tố: Ung thư da không phải u hắc tố (NMSC), bao gồm ung thư tế bào đáy, ung thư tế bào vảy và ung thư biểu mô tế bào Merkel đã được báo cáo ở những bệnh nhân điều trị bằng Jakavi. Hầu hết những bệnh nhân này có tiền sử điều trị kéo dài bằng hydroxyurea và bị NMSC trước đó hoặc có tổn thương da tiền ác tính. Mối quan hệ nhân quả với ruxolitinib chưa được xác định. Khuyến cáo nên khám da định kỳ cho những bệnh nhân có nguy cơ cao về ung thư da. – Các nhóm bệnh nhân đặc biệtSuy thận: Nên giảm liều khởi đầu của Jakavi ở bệnh nhân suy thận nặng. Đối với bệnh nhân bị bệnh thận giai đoạn cuối đang được thẩm phân, liều khởi đầu nên dựa trên số lượng tiểu cầu. Chỉ nên dùng các liều kế tiếp đối với bệnh nhân vào những ngày lọc thận sau mỗi lần lọc thận. Việc điều chỉnh liều thêm nên dựa vào độ an toàn và hiệu quả của thuốc. Suy gan: Nên giảm liều khởi đầu của Jakavi ở bệnh nhân suy gan. Việc điều chỉnh liều thêm nên dựa trên độ an toàn và hiệu quả của thuốc. Tương tác thuốc: Nếu Jakavi được dùng đồng thời với thuốc ức chế mạnh CYP3A4 hoặc thuốc ức chế kép trung bình CYP2C9 va CYP3A4 (ví dụ: fluconazol), nên giảm liều khoảng 50%. Các tác dụng do ngừng thuốc: Sau khi ngừng điều trị, các triệu chứng liên quan với xơ hóa tủy xương có khả năng quay trở lại. – Thời kỳ mang thai và cho con búPhụ nữ có khả năng mang thai: Những phụ nữ có khả năng mang thai phải có biện pháp phòng ngừa thích hợp để tránh mang thai trong khi điều trị. Trong trường hợp mang thai xảy ra, phải thực hiện đánh giá nguy cơ/lợi ích trên cơ sở từng bệnh nhân với sự tư vấn cẩn thận về nguy cơ tiềm ẩn đối với thai bằng cách sử dụng các dữ liệu cập nhật nhất. Phụ nữ có thai: Chưa có các nghiên cứu đầy đủ và đối chứng chặt chẽ về Jakavi ở phụ nữ có thai. Các nghiên cứu về sự phát triển của phôi thai với ruxolitinib ở chuột cống và chuột nhắt không cho thấy tính gây quái thai. Ruxolitinib có độc tính đối với phôi và độc tính đối với thai ở chuột cống (tăng mất phôi sau khi làm tổ và giảm cân nặng của thai). Chưa rõ nguy cơ tiềm ẩn đối với người. Không khuyến cáo sử dụng Jakavi trong khi mang thai. Phụ nữ cho con bú: Phụ nữ đang dùng Jakavi không nên cho con bú. Ở chuột cống cho con bú, ruxolitinib và/hoặc các chất chuyển hóa của nó được bài tiết vào sữa với nồng độ cao hơn 13 lần so với nồng độ trong huyết tương ở chuột mẹ. Chưa rõ liệu Jakavi có bài tiết vào sữa mẹ hay không. Khả năng sinh sản: Chưa có dữ liệu ở người về tác dụng của ruxolitinib trên khả năng sinh sản. Trong các nghiên cứu ở động vật, không quan sát thấy các tác dụng có hại trên khả năng sinh sản hoặc hiệu suất sinh sản ở chuột cống đực hoặc cái. Trong một nghiên cứu trước sinh và sau sinh ở chuột cống, khả năng sinh sản ở chuột con thế hệ đầu tiên cũng không bị ảnh hưởng. – Tương tác thuốcCác tác nhân làm thay đổi nồng độ ruxolitinib trong huyết tương Thuốc ức chế mạnh CYP3A4:
Thuốc ức chế nhẹ hoặc trung bình CYP3A4:
Thuốc ức chế kép trung bình CYP2C9 và CYP3A4 (ví dụ fluconazole):
Thuốc gây cảm ứng CYP3A4:
Ảnh hưởng của ruxolitinib đến các thuốc khác Các chất được vận chuyển bởi P-glycoprotein hoặc các chất vận chuyển khác:
Các yếu tố tăng trưởng tạo máu:
Điều trị hóa chất làm giảm tế bào:
Cơ chất của CYP3A4:
Thuốc tránh thai dùng đường uống:
|
7. Dược lý
– Cơ chế tác dụng (MOA)
Ruxolitinib là một thuốc ức chế chọn lọc Janus Associated Kinase (JAK) JAK1 và JAK2 (trị số IC50 là 3,3 nM đối với enzym JAK1 và 2,8 nM đối với enzym JAK2). Các enzym này làm trung gian truyền tín hiệu của một số cytokin và hormon tăng trưởng quan trọng đối với chức năng tạo máu và miễn dịch. Sự truyền tín hiệu qua con đường JAK liên quan đến sự huy động STAT (yếu tố chuyển đổi tín hiệu và yếu tố hoạt hóa sự phiên mã) cho các thụ thể cytokine, sự hoạt hóa và sự phân bổ khu trú sau đó của STAT ở nhân dẫn đến điều hòa biểu hiện gen. Rối loạn điều hòa con đường JAK – STAT có liên quan với một số ung thư, gia tăng sự tăng sinh và sự sống sót của các tế bào ác tính.
Xơ tủy xương (MF) là một rối loạn tăng sinh tủy xương (MPN) đã biết có liên quan với sự rối loạn điều hòa việc truyền tín hiệu qua con đường JAK1 và JAK2. Cơ sở về rối loạn điều hòa được cho là bao gồm nồng độ cao của cytokine trong tuần hoàn hoạt hóa con đường JAK – STAT, các đột biến tăng chức năng như JAK2V617F và ức chế các cơ chế điều hòa âm tính. Những bệnh nhân bị xơ hóa tủy xương biểu hiện rối loạn điều hòa dẫn truyền tín hiệu qua con đường JAK bất kể tình trạng đột biến JAK2V617F.
Ruxolitinib ức chế sự truyền tín hiệu qua con đường JAK – STAT và sự tăng sinh tế bào trong các mô hình tế bào của các u ác tính về huyết học phụ thuộc cytokine, cũng như của tế bào Ba/F3 làm cho chúng không phụ thuộc cytokine bằng cách biểu hiện protein đột biến JAK2V617F, với trị số IC50 trong khoảng từ 80 – 320 nM. Trong một mô hình u tăng sinh tủy xương với JAK2V617F dương tính, sử dụng ruxolitinib đường uống đã ngăn ngừa lách to, ưu tiên làm giảm các tế bào đột biến JAK2V617F ở lách, làm giảm các cytokine gây viêm trong tuần hoàn (ví dụ TNF-a, IL-6) và dẫn đến kéo dài sự sống còn đáng kể ở chuột nhắt ở các liều không gây tác dụng ức chế tủy xương.
– Dược lực học
Ruxolitinib ức chế sự phosphoryl hóa STAT3 gây ra bởi cytokine trong máu toàn phần ở các đối tượng khỏe mạnh và bệnh nhân bị xơ tủy xương. Ruxolitinib dẫn đến ức chế tối đa sự phosphoryl hóa STAT3 sau khi dùng thuốc 2 giờ, trở về gần mức ban đầu lúc 8 giờ ở cả đối tượng khỏe mạnh và bệnh nhân bị xơ tủy xương, điều này cho thấy không có sự tích lũy thuốc ban đầu hoặc các chất chuyển hóa có hoạt tính.
Sự tăng ban đầu về các yếu tố chỉ điểm viêm có liên quan với các triệu chứng toàn thân như TNFalpha, IL-6 và CRP ở các đối tượng bị xơ tủy xương giảm xuống sau khi điều trị bằng ruxolitinib. Bệnh nhân bị xơ tủy xương không bị đề kháng với các tác dụng dược lực học của việc điều trị bằng ruxolitinib theo thời gian.
Trong một nghiên cứu kỹ lưỡng về khoảng QT ở các đối tượng khỏe mạnh, không có dấu hiệu về tác dụng kéo dài khoảng QT/QTc do ruxolitinib ở các liều đơn lên đến liều 200 mg cao vượt liều điều trị, điều này cho thấy ruxolitinib không có ảnh hưởng đến sự tái cực của tim.
– Dược động học
Hấp thu:
Ruxolitinib là một phân tử Loại 1 theo Hệ thống Phân loại Dược phẩm sinh học, có tính thấm cao, độ hòa tan cao và đặc tính hòa tan nhanh. Trong các nghiên cứu lâm sàng, ruxolitinib được hấp thu nhanh sau khi dùng đường uống với nồng độ tối đa trong huyết tương (Cmax) đạt được khoảng 1 giờ sau khi dùng thuốc. Dựa trên nghiên cứu về cân bằng khối lượng ở người, sự hấp thu ruxolitinib qua đường uống là 95% hoặc cao hơn. Cmax trung bình của ruxolitinib và nồng độ tổng cộng (diện tích dưới đường cong, AUC) tăng tỉ lệ thuận trong phạm vi liều đơn từ 5 – 200 mg. Không có sự thay đổi nào có ý nghĩa lâm sàng về dược động học của ruxolitinib khi dùng với bữa ăn nhiều chất béo. Cmax trung bình giảm vừa phải (24%) trong khi AUC trung bình gần như không thay đổi (tăng 4%) khi dùng thuốc với bữa ăn nhiều chất béo.
Phân bố:
Thể tích phân bố trung bình ở trạng thái ổn định khoảng 75 lít ở bệnh nhân bị xơ tủy xương. Ở các nồng độ của ruxolitinib có ý nghĩa lâm sàng, sự gắn kết với protein huyết tương in vitro khoảng 97%, chủ yếu là với albumin. Một nghiên cứu chụp xạ hình toàn cơ thể ở chuột cống đã cho thấy ruxolitinib không thấm qua hàng rào máu não.
Biến đổi sinh học/chuyển hóa:
Các nghiên cứu in vitro cho thấy CYP3A4 là một enzym chính chịu trách nhiệm về sự chuyển hóa ruxolitinib. Hợp chất ban đầu là thực thể chủ yếu ở người, chiếm khoảng 60% của chất liên quan đến thuốc trong tuần hoàn. Hai chất chuyển hóa chính và có hoạt tính được xác định trong huyết tương của các đối tượng khỏe mạnh, đại diện 25% và 11% về AUC của thuốc ban đầu. Những chất chuyển hóa này có một nửa đến 1/5 hoạt tính dược lý liên quan với JAK của thuốc ban đầu. Tổng tất cả các chất chuyển hóa có hoạt tính đóng góp 18% vào dược lực học của ruxolitinib về tổng thể. Ở các nồng độ có ý nghĩa lâm sàng, ruxolitinib không ức chế CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 hoặc CYP3A4 và không phải là thuốc gây cảm ứng mạnh CYP1A2, CYP2B6 hoặc CYP3A4 dựa trên các nghiên cứu in vitro.
Thải trừ:
Sau khi cho các đối tượng người lớn khỏe mạnh uống một liều đơn ruxolitinib có đánh dấu phóng xạ (14C), sự thải trừ chủ yếu thông qua chuyển hóa với 74% hoạt tính phóng xạ được bài tiết trong nước tiểu và 22% thải trừ qua phân. Lượng thuốc dạng không biến đổi chiếm dưới 1% tổng hoạt tính phóng xạ được bài tiết. Thời gian bán thải trung bình của ruxolitinib khoảng 3 giờ.
Sự tuyến tính/không tuyến tính
Sự tỷ lệ thuận với liều dùng đã được chứng minh trong các nghiên cứu đơn liều và đa liều.
Các nhóm đối tượng đặc biệt
Ảnh hưởng của tuổi, giới tính hoặc chủng tộc:
Dựa trên các nghiên cứu ở các đối tượng khỏe mạnh, không quan sát thấy sự khác biệt có ý nghĩa về dược động học của ruxolitinib về mặt giới tính và chủng tộc. Trong một đánh giá dược động học quần thể trên bệnh nhân bị xơ tủy xương, không có mối quan hệ nào rõ ràng giữa độ thanh thải đường uống và tuổi hoặc chủng tộc của bệnh nhân. Độ thanh thải là 17,7 lít/giờ ở phụ nữ và 22,1 lít/giờ ở nam giới, với độ biến thiên 39% giữa các đối tượng nghiên cứu.
Bệnh nhi:
Độ an toàn và hiệu quả của Jakavi ở bệnh nhi chưa được xác định.
Suy thận:
Sau khi dùng một liều đơn ruxolitinib 25 mg, dược động học tương tự nhau ở các đối tượng có các mức độ suy thận khác nhau và ở những người có chức năng thận bình thường. Tuy nhiên, các trị số AUC của các chất chuyển hóa của ruxolitinib trong huyết tương có xu hướng tăng theo mức độ nặng của suy thận tăng lên và rõ rệt nhất ở các đối tượng bị bệnh thận giai đoạn cuối cần phải thẩm phân máu. Ruxolitinib không bị loại bỏ bằng thẩm phân. Khuyến cáo điều chỉnh liều đối với bệnh nhân bị suy thận nặng (độ thanh thải creatinin (Clcr) dưới 30 ml/phút). Đối với những bệnh nhân bị bệnh thận giai đoạn cuối, khuyến cáo điều chỉnh phác đồ dùng thuốc.
Suy gan:
Sau khi dùng một liều đơn ruxolitinib 25 mg cho những bệnh nhân có các mức độ suy gan khác nhau, dược động học và dược lực học của ruxolitinib đã được đánh giá. AUC trung bình của ruxolitinib tăng ở bệnh nhân suy gan nhẹ là 87%, ở bệnh nhân suy gan trung bình là 28% và ở bệnh nhân suy gan nặng là 65%, so với bệnh nhân có chức năng gan bình thường và cho thấy không có mối quan hệ rõ ràng với mức độ suy gan dựa trên điểm số Child – Pugh. Thời gian bán thải cuối cùng kéo dài ở bệnh nhân suy gan so với nhóm đối chứng khỏe mạnh (4,1 – 5,0 giờ so với 2,8 giờ). Khuyến cáo giảm liều đối với bệnh nhân suy gan.
– Các nghiên cứu lâm sàng
Hai nghiên cứu lâm sàng ngẫu nhiên pha 3 (COMFORT-I và COMFORT-II) đã được tiến hành trên bệnh nhân bị xơ tủy xương (xơ tủy nguyên phát, xơ tủy sau đa hồng cầu vô căn hoặc xơ tủy sau tăng tiểu cầu vô căn). Trong cả hai nghiên cứu, bệnh nhân có lách to sờ thấy được ít nhất là 5 cm dưới bờ sườn và thuộc nhóm nguy cơ trung bình 2 (có 2 yếu tố tiên lượng) hoặc nguy cơ cao (có 3 yếu tố tiên lượng trở lên) dựa trên Tiêu chí Đồng thuận của Nhóm Nghiên cứu Quốc tế (IWG). Các yếu tố tiên lượng bao gồm các tiêu chí theo IWG gồm tuổi > 65, có các triệu chứng toàn thân (giảm cân, sốt, ra mồ hôi đêm), thiếu máu (hemoglobin < 10 g/dl), tăng bạch cầu (tiền sử có số lượng bạch cầu (WBC) > 25 x 109/l) và nguyên bào trong máu ≥ 1%. Liều khởi đầu của Jakavi được dựa trên số lượng tiểu cầu. Những bệnh nhân có số lượng tiểu cầu từ 100.000 – 200.000/mm3 được điều trị khởi đầu bằng Jakavi 15 mg, 2 lần/ngày và những bệnh nhân có số lượng tiểu cầu > 200.000/mm3 được điều trị khởi đầu bằng Jakavi 20 mg, 2 lần/ngày. Sau đó các liều được điều chỉnh theo từng bệnh nhân dựa trên khả năng dung nạp và hiệu quả với liều tối đa là 20 mg, 2 lần/ngày đối với những bệnh nhân có số lượng tiểu cầu từ 100.000 đến ≤ 125.000/mm3, liều 10 mg, 2 lần/ngày đối với những bệnh nhân có số lượng tiểu cầu từ 75.000 đến ≤ 100.000/mm3, và liều 5 mg, 2 lần/ngày đối với những bệnh nhân có số lượng tiểu cầu từ 50.000 đến ≤ 75.000/mm3.
COMFORT-I là một nghiên cứu mù đôi, ngẫu nhiên, có đối chứng với giả dược ở 309 bệnh nhân kháng trị hoặc không phải là đối tượng cho trị liệu hiện có. Các bệnh nhân được cho dùng Jakavi hoặc giả dược đối chứng. Tiêu chí đánh giá chính là tỷ lệ các đối tượng đạt được mức giảm ≥ 35% so với lúc ban đầu về thể tích của lách ở tuần thứ 24 khi được đánh giá bằng chụp cộng hưởng từ (MRI) hoặc chụp cắt lớp điện toán (CT).
Các tiêu chí phụ bao gồm thời gian duy trì mức giảm ≥ 35% so với lúc ban đầu về thể tích của lách, tỷ lệ bệnh nhân có mức giảm ≥ 50% về tổng điểm số triệu chứng từ lúc ban đầu đến tuần thứ 24 khi được đánh giá bằng nhật ký triệu chứng phiên bản 2,0 theo Mẫu đánh giá Triệu chứng Xơ Tủy xương (MFSAF) sửa đổi, thay đổi về tổng điểm số triệu chứng từ lúc ban đầu đến tuần thứ 24 khi được đánh giá bằng nhật ký triệu chứng phiên bản 2.0 theo MFSAF và sống còn toàn bộ.
COMFORT-II là một nghiên cứu nhãn mỡ, ngẫu nhiên ở 219 bệnh nhân. Các bệnh nhân được chọn ngẫu nhiên 2:1 điều trị bằng Jakavi so với trị liệu tốt nhất hiện có. Trị liệu tốt nhất hiện có được lựa chọn bởi nhà nghiên cứu trên từng bệnh nhân. Ở nhóm dùng trị liệu tốt nhất hiện có, 47% bệnh nhân được điều trị bằng hydroxyurea và 16% bệnh nhân được điều trị bằng glucocorticoid. Tiêu chí đánh giá chính là tỷ lệ bệnh nhân đạt được mức giảm ≥ 35% so với lúc ban đầu về thể tích của lách ở tuần thứ 48 khi được đánh giá bằng chụp MRI hoặc CT.
Một tiêu chí phụ trong nghiên cứu COMFORT-II là tỷ lệ bệnh nhân đạt được mức giảm ≥ 35% về thể tích của lách khi được đánh giá bằng chụp MRI hoặc CT từ lúc ban đầu đến tuần thứ 24. Thời gian duy trì mức giảm ≥ 35% so với lúc ban đầu ở bệnh nhân đáp ứng cũng là một tiêu chí phụ.
Trong nghiên cứu COMFORT-I, đặc điểm bệnh nhân lúc ban đầu và đặc điểm về bệnh ở bệnh nhân tương tự giữa 2 nhóm điều trị. Tuổi trung vị là 68 với 61% bệnh nhân trên 65 tuổi và 54% nam. 50% bệnh nhân bị xơ tủy xương nguyên phát, 31% bị xơ tủy sau đa hồng cầu vô căn và 18% bị xơ tủy sau tăng tiểu cầu vô căn. 21% bệnh nhân có các đợt truyền hồng cầu trong vòng 8 tuần tham gia vào nghiên cứu. Số lượng tiểu cầu trung vị là 251.000/mm3. 76% bệnh nhân có đột biến mã hóa thay thế V617F có trong protein JAK. Các bệnh nhân có chiều dài lách trung vị sờ thấy được là 16 cm. Lúc ban đầu, 37,4% bệnh nhân ở nhóm Jakavi bị thiếu máu độ 1; 31,6% độ 2 và 4,5% độ 3, trong khi ở nhóm giả dược 35,8% bị thiếu máu độ 1; 35,1% độ 2; 4,6% độ 3 và 0,7% độ 4. Giảm tiểu cầu độ 1 được tìm thấy ở 12,9% bệnh nhân trong nhóm Jakavi và 13,2% ở nhóm giả dược.
Trong nghiên cứu COMFORT-II, đặc điểm bệnh nhân lúc ban đầu và đặc điểm về bệnh ở bệnh nhân tương tự giữa 2 nhóm điều trị. Tuổi trung vị là 66 với 52% bệnh nhân trên 65 tuổi và 57% nam. 53% bệnh nhân bị xơ tủy nguyên phát, 31% bị xơ tủy sau đa hồng cầu vô căn và 16% bị xơ tủy sau tăng tiểu cầu vô căn. 19% bệnh nhân được xem là phụ thuộc vào truyền máu lúc ban đầu. Các bệnh nhân có chiều dài lách trung vị sờ thấy được là 15 cm.
Lúc ban đầu, 34,2% bệnh nhân ở nhóm Jakavi bị thiếu máu độ 1; 28,8% độ 2 và 7,5% độ 3, trong khi ở nhóm dùng trị liệu tốt nhất hiện có thì 37% bị thiếu máu độ 1; 27,4% độ 2, 13,7% độ 3 và 1,4% độ 4. Giảm tiểu cầu độ 1 được phát hiện ở 8,2% bệnh nhân ở nhóm Jakavi và 9,6% ở nhóm dùng trị liệu tốt nhất hiện có. Phân tích hiệu quả theo tiêu chỉ đánh giá chính trong các nghiên cứu COMFORT-I và COMFORT-II được trình bày ở Bảng 2 dưới đây. Một tỷ lệ bệnh nhân cao hơn đáng kể ở nhóm Jakavi đạt được mức giảm ≥ 35% về thể tích của lách so với lúc ban đầu ở cà hai nghiên cứu so với nhóm giả dược trong nghiên cứu COMFORT-I và trị liệu tốt nhất hiện có trong nghiên cứu COMFORT-II.
Bảng 2. Tỷ lệ phần trăm bệnh nhân có mức giảm ≥ 35% so với lúc ban đầu về thể tích của lách ở tuần thứ 24 trong nghiên cứu COMFORT-I và ở tuần thứ 48 trong nghiên cứu COMFORT-II (theo ý định điều trị – ITT).
| COMFORT-l | COMFORT-ll | ||
| Jakavi (N = 155) | Giả dược (N = 153) | Jakavi (N = 144) | Trị liệu tốt nhất hiện có (N = 72) |
Thời điểm | Tuần thứ 24 | Tuần thứ 48 | ||
Số đối tượng (%) có mức giảm thể tích của lách ≥ 35% | 65 (41,9) | 1 (0,7) | 41 (28,5) | 0 |
Khoảng tin cậy 95% | 34,1 – 50,1 | 0 – 3,6 | 21,3 – 36,6 | 0,0 – 5,0 |
Giá trị P | < 0,0001 | < 0,0001 | ||
Trong nghiên cứu COMFORT-I; 41,9% bệnh nhân ở nhóm Jakavi đạt được mức giảm ≥ 35% về thể tích của lách so với lúc ban đầu so với 0,7% ở nhóm giả dược vào tuần thứ 24. Một tỷ lệ bệnh nhân tương tự ở nhóm Jakavi đạt được mức giảm ≥ 50% về chiều dài của lách sở thấy được.
Trong nghiên cứu COMFORT-II; 28,5% bệnh nhân ở nhóm Jakavi đạt được mức giảm ≥ 35% về thể tích của lách so với lúc ban đầu so với 0 bệnh nhân (0%) ở nhóm dùng trị liệu tốt nhất hiện có vào tuần thứ 48. Một tiêu chí phụ là tỷ lệ bệnh nhân đạt được mức giảm ≥ 35% về thể tích của lách ở tuần thứ 24. Một tỷ lệ bệnh nhân cao hơn có ý nghĩa ở nhóm Jakavi là 46 bệnh nhân (31,9%) đạt được mức giảm ≥ 35% về thể tích của lách so với lúc ban đầu, so với 0 bệnh nhân (0%) ở nhóm dùng trị liệu tốt nhất hiện có (giá trị p < 0,0001).
Một tỷ lệ bệnh nhân cao hơn đáng kể ở nhóm Jakavi đạt được mức giảm ≥ 35% so với lúc ban đầu về thể tích của lách bất kể có hoặc không có đột biến JAK2V617F hoặc subtype của bệnh (xơ tủy xương nguyên phát, xơ tủy sau đa hồng cầu vô căn hoặc xơ tủy sau tăng tiểu cầu vô căn).
Hình 1 cho thấy một biểu đồ dạng thác nước về thay đổi phần trăm so với lúc ban đầu về thể tích của lách ở tuần thứ 24 trong nghiên cứu COMFORT-I. Trong số 139 bệnh nhân ở nhóm Jakavi có cả hai đánh giá về thể tích của lách lúc ban đầu và ở tuần thứ 24, tất cả trừ 2 bệnh nhân có giảm ở mức nhất định về thể tích của lách ở tuần thứ 24, với mức giảm trung bình là 33%. Trong số 106 bệnh nhân ở nhóm giả dược có cả hai đánh giá về thể tích của lách lúc ban đầu và ở tuần thứ 24, có sự tăng trung vị là 8,5%.
Hình 1 Biểu đồ thác nước về thay đổi phần trăm so với lúc ban đầu về thể tích của lách ở tuần thứ 24 (Các trường hợp được quan sát) COMFORT-I
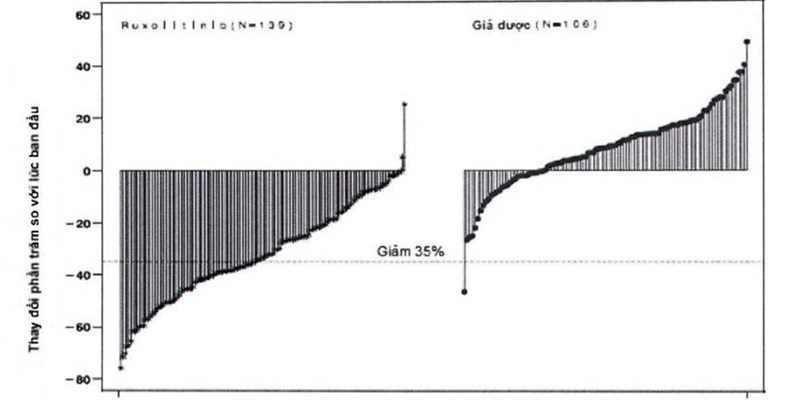
Hình 2 trình bày một biểu đồ thác nước về thay đổi phần trăm so với lúc ban đầu về thể tích của lách ở tuần thứ 48 trong nghiên cứu COMFORT-II. Trong số 98 bệnh nhân ở nhóm Jakavi có cả hai đánh giá về thể tích của lách lúc ban đầu và ở tuần thứ 48, mức giảm trung vị về thể tích của lách ở tuần thứ 48 là 28%. Trong số 34 bệnh nhân ở nhóm dùng trị liệu tốt nhất hiện có là những bệnh nhân có cả hai đánh giá về thể tích của lách lúc ban đầu và ở tuần thứ 48, có sự tăng trung vị là 8,5%.
Hình 2. Biểu đồ thác nước về thay đổi phần trăm so với lúc ban đầu về thể tích của lách ở tuần thứ 48 trong nghiên cứu COMFORT- II
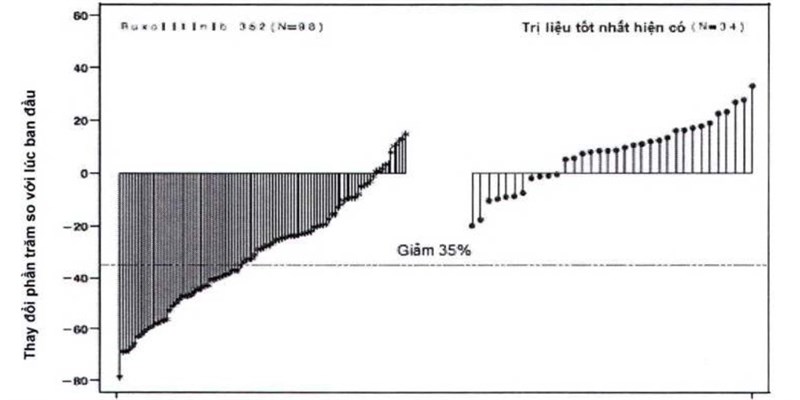
Xác suất liên quan thời gian từ mức giảm đầu tiên ≥ 35% về thể tích của lách đến khi tăng 25% so với mức giảm thấp nhất và mất đáp ứng trong nghiên cứu COMFORT-I và COMFORT-II được trình bày trong Bảng 3 dưới đây.
Bảng 3. Phân tích Kaplan – Meier về duy trì khoảng thời gian từ mức giảm đầu tiên ≥ 35% về thể tích của lách đến tăng 25% so với mức thấp nhất và mất đáp ứng ở những bệnh nhân dùng Jakavi (COMFORT-I và COMFORT-II)
Thống kê | Jakavi (COMFORT-l) | Jakavi (COMFORT-ll) |
Xác suất duy trì khoảng thời gian > 12 tuần (khoảng tin cậy (Cl) 95%) | 0,98 (0,89 – 1,00) | 0,92 (0,82 – 0,97) |
Xác suất duy trì khoảng thời gian > 24 tuần (khoảng tin cậy 95%) | 0,89 (0,75 – 0,95) | 0,87 (0,76 – 0,93) |
Xác suất duy trì khoảng thời gian > 36 tuần (khoảng tin cậy 95%) | 0,71 (0,41 – 0,88) | 0,77 (0,63 – 0,87) |
Xác suất duy trì khoảng thời gian > 48 tuần (khoảng tin cậy 95%) | Không áp dụng | 0,52 (0,18 – 0,78) |
Trong số 80 bệnh nhân có mức giảm ≥ 35% ở bất kỳ thời điểm nào trong nghiên cứu COMFORT-I và trong số 69 bệnh nhân ở nghiên cứu COMFORT-II, xác suất một bệnh nhân duy trì đáp ứng khi đang dùng Jakavi trong ít nhất 24 tuần là 89% trong nghiên cứu COMFORT-I và 87% trong nghiên cứu COMFORT-II và xác suất duy trì đáp ứng trong ít nhất 48 tuần là 52% trong nghiên cứu COMFORT-II.
Jakavi cải thiện các triệu chứng liên quan xơ tủy và cải thiện chất lượng cuộc sống (QOL) ở những bệnh nhân bị xơ tủy nguyên phát (PMF), xơ tủy sau đa hồng cầu vô căn (PPV – MF) hoặc xơ tủy sau tăng tiểu cầu vô căn (PET – MF). Trong nghiên cứu COMFORT-I, các triệu chứng xơ tủy được ghi nhận sử dụng nhật ký triệu chứng phiên bản 2.0 theo MFSAF sửa đổi dưới dạng nhật ký điện tử được các đối tượng hoàn tất hàng ngày. Thay đổi so với lúc ban đầu về tổng điểm số triệu chứng ở tuần thứ 24 là tiêu chí phụ trong nghiên cứu này. Một tỷ lệ bệnh nhân cao hơn có ý nghĩa ở nhóm Jakavi đã đạt được sự cải thiện 2 50% so với lúc ban đầu về tổng điểm số triệu chứng ở tuần thứ 24 so với nhóm giả dược (45,9% ở nhóm Jakavi và 5,3% ở nhóm giả dược, p < 0,0001 sử dụng kiểm định chi bình phương).
Sự cải thiện chất lượng cuộc sống về tổng thể đã được đánh giá bằng Bảng câu hỏi chất lượng cuộc sống của Tổ chức Nghiên cứu và Điều trị Ung thư Châu Âu (EORTC QLQ-C30) trong cả hai nghiên cứu COMFORT-I và COMFORT-II.
COMFORT-I so sánh Jakavi với giả dược lúc 24 tuần và COMFORT-II so sánh Jakavi với trị liệu tốt nhất hiện có lúc 48 tuần. Vào lúc ban đầu đối với cả hai nghiên cứu, điểm số theo từng thang điểm phụ của thang điểm đánh giá EORTC QLQ-C30.
Đối với nhóm Jakavi và nhóm dùng thuốc so sánh là tương tự nhau. Vào tuần thứ 24 ở nghiên cứu COMFORT-I, nhóm Jakavi cho thấy sự cải thiện có ý nghĩa về tình trạng sức khỏe chung/chất lượng cuộc sống theo thang điểm đánh giá EORTC QLQ-C30 so với nhóm giả dược (thay đổi trung bình +12,3 đối với nhóm Jakavi và – 3,4 đối với nhóm giả dược, p < 0,0001). Ở tuần thứ 24 và tuần thứ 48, nhóm Jakavi ở nghiên cứu COMFORT-II cho thấy xu hướng cải thiện nhiều hơn về tiêu chí thăm dò là tình trạng sức khỏe chung/chất lượng cuộc sống so với trị liệu tốt nhất hiện có và điều này phù hợp với các kết quả trong nghiên cứu COMFORT-I.
Trong nghiên cứu COMFORT-I, sau một thời gian theo dõi trung vị là 34,3 tháng, tỷ lệ tử vong ở bệnh nhân được phân ngẫu nhiên dùng ruxolitinib là 27,1% (42 trong số 155 bệnh nhân) so với 35,1% (54 trong số 154 bệnh nhân) thuộc nhóm dùng giả dược. Ruxolitinib làm giảm nguy cơ tử vong 31,3% so với giả dược (HR 0,687; KTC 95% 0,459 – 1,029; p = 0,0668).
Trong nghiên cứu COMFORT-II, sau một thời gian theo dõi trung vị là 34,7 tháng, tỷ lệ tử vong ở bệnh nhân được phân ngẫu nhiên dùng ruxolitinib là 19,9% (29 trong số 146 bệnh nhân) so với 30,1% (22 trong số 73 bệnh nhân) thuộc nhóm được điều trị với liệu pháp tốt nhất có được (BAT: best available therapy). Ruxolitinib làm giảm nguy cơ tử vong 52% so với nhóm BAT (HR 0,48; KTC 95% 0,28 – 0,85; p = 0,009).
8. Thông tin chung
– Đặc điểm
Viên nén.
– Bảo quản
Bảo quản nơi khô ráo, thoáng mát, ở nhiệt độ không quá 30°C.
Giữ thuốc trong bao bì gốc. Không dùng Jakavi quá hạn sử dụng được ghi “EXP” trên bao bì.
– Quy cách đóng gói
Hộp 4 vỉ x 14 viên.
– Hạn dùng
24 tháng kể từ ngày sản xuất.
– Nhà sản xuất
Novartis Pharma Stein AG.



Đánh giá
Chưa có đánh giá nào.